

 首页
首页 关于我们
关于我们 产品中心
产品中心 技术服务
技术服务 技术中心
技术中心 联系我们
联系我们
导读
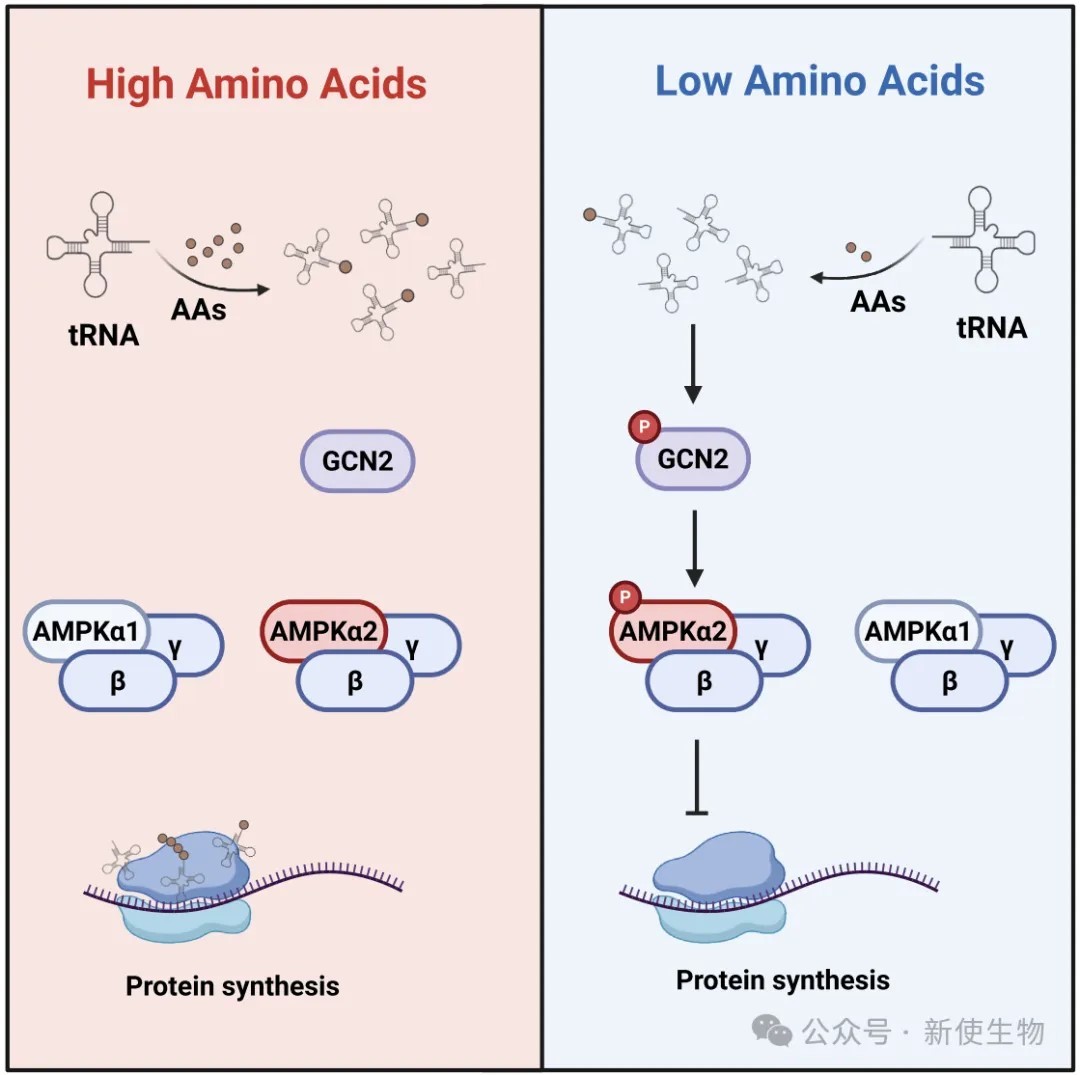
AMPK是一种关键的能量感受器,由一个催化亚基(α)和两个调节亚基(β, γ)组成。其催化亚基存在α1和α2两种高度相似的同工酶,但它们在功能上的具体分工尚不明确。
AMPK的激活依赖于其α亚基上苏氨酸172位点(T172)的磷酸化,该过程主要响应低能量状态(如高AMP/ATP比值)。蛋白质合成是一个高耗能过程,需要充足的氨基酸作为原料。
GCN2激酶是细胞内感受氨基酸缺乏的经典传感器。然而,AMPK是否能直接感受氨基酸水平的变化,并以此来调控蛋白质合成,特别是α1和α2亚基在此过程中是否扮演不同角色,仍是未解之谜。
2025年10月7日,复旦大学赵世民/徐薇/陈兴栋团队合作,在Cell Metabolism上发表了题为“AMPKα2 signals amino acid insufficiency to inhibit protein synthesis”的论文。该研究颠覆了AMPK主要作为能量感受器的传统认知,首次揭示了其催化亚基AMPKα2(而非α1)是氨基酸匮乏信号的特异性感受器。

文章索引
【标题】AMPKα2 signals amino acid insufficiency to inhibit protein synthesis
【发表期刊】Cell Metabolism
【发表日期】2025年10月7日
【作者及团队】复旦大学赵世民/陈兴栋/徐薇团队
【IF】30.9
研究结果
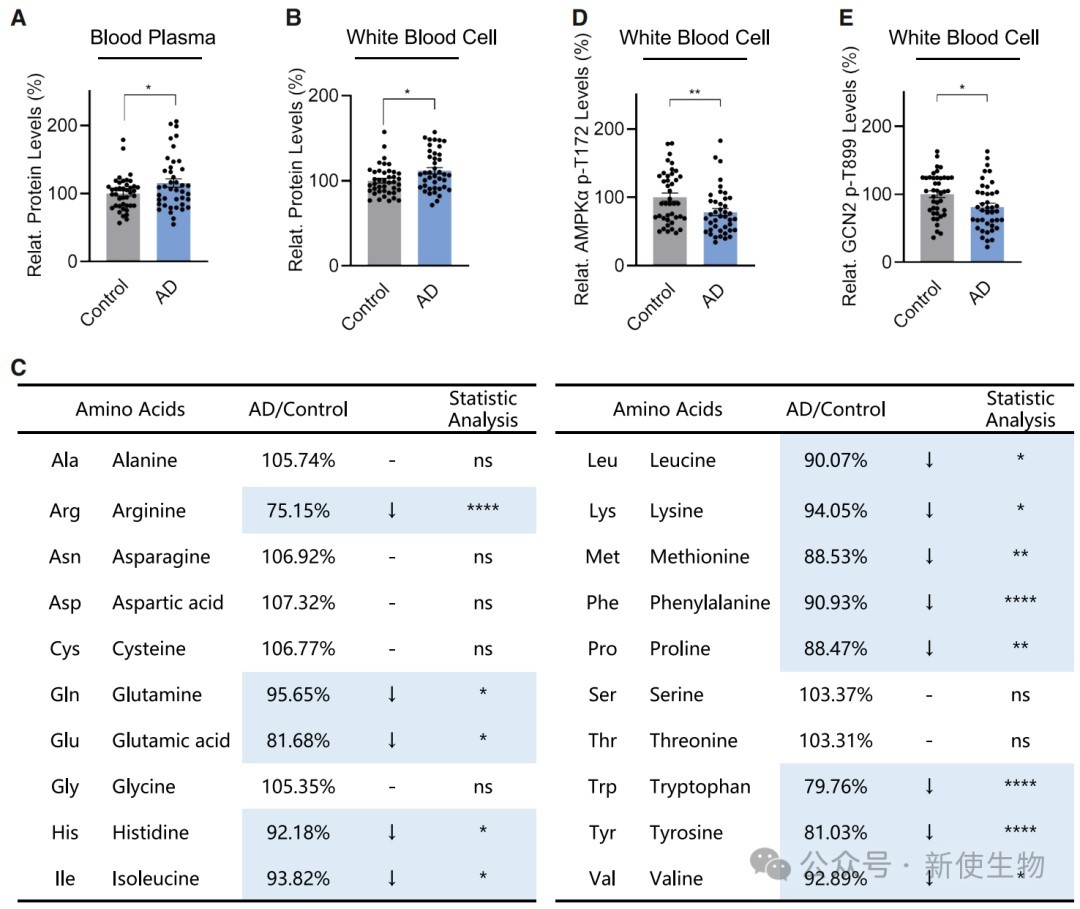
一、AD患者血浆中存在蛋白质合成失调的迹象
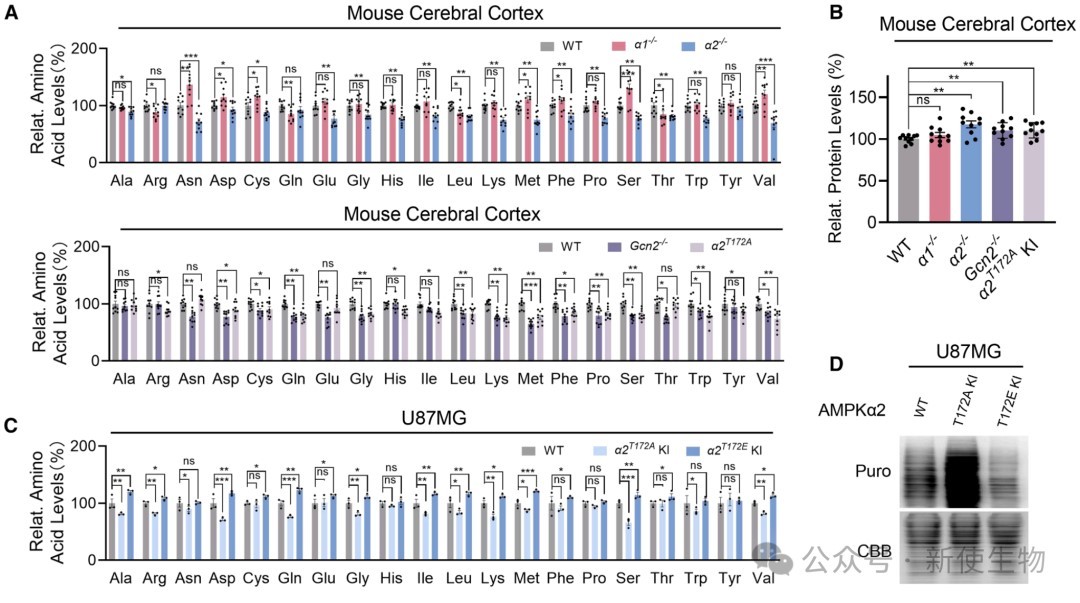
通过对一个大型中国人群队列的AD患者样本进行分析,研究团队发现与健康对照组相比,AD患者血浆和白细胞中总蛋白水平升高,而多种氨基酸水平下降。
同时,负责抑制蛋白质合成的关键激酶AMPKα和GCN2的磷酸化激活水平均显著降低。
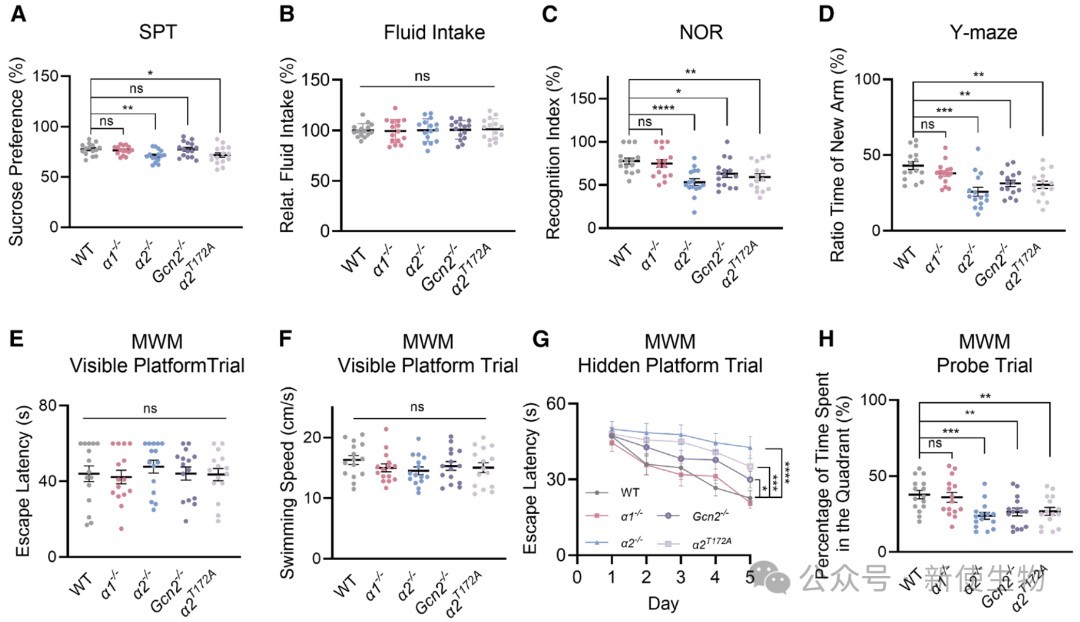
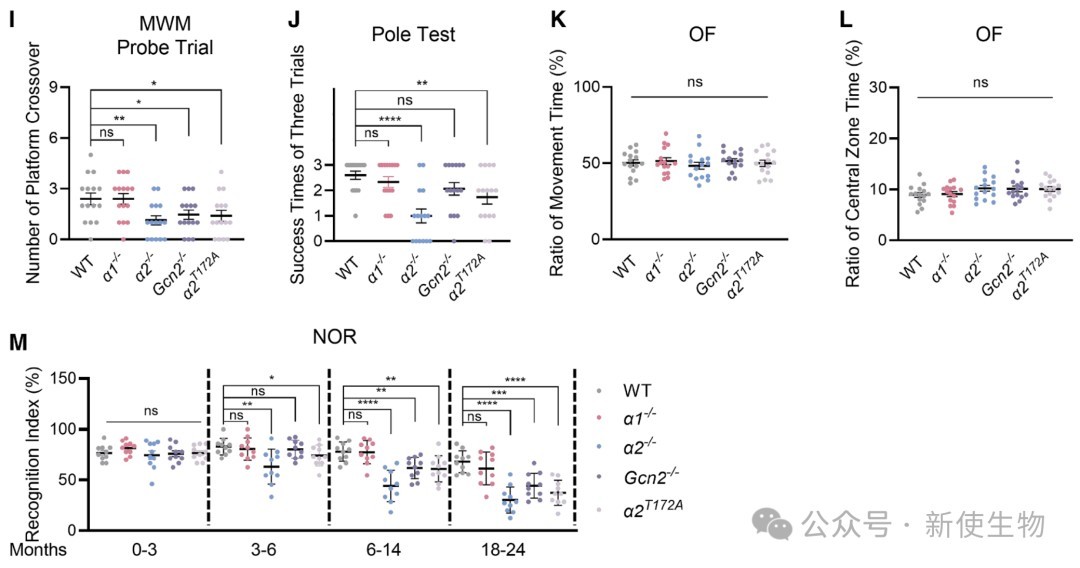
二、AMPKα2而非α1的缺失会导致AD样表型
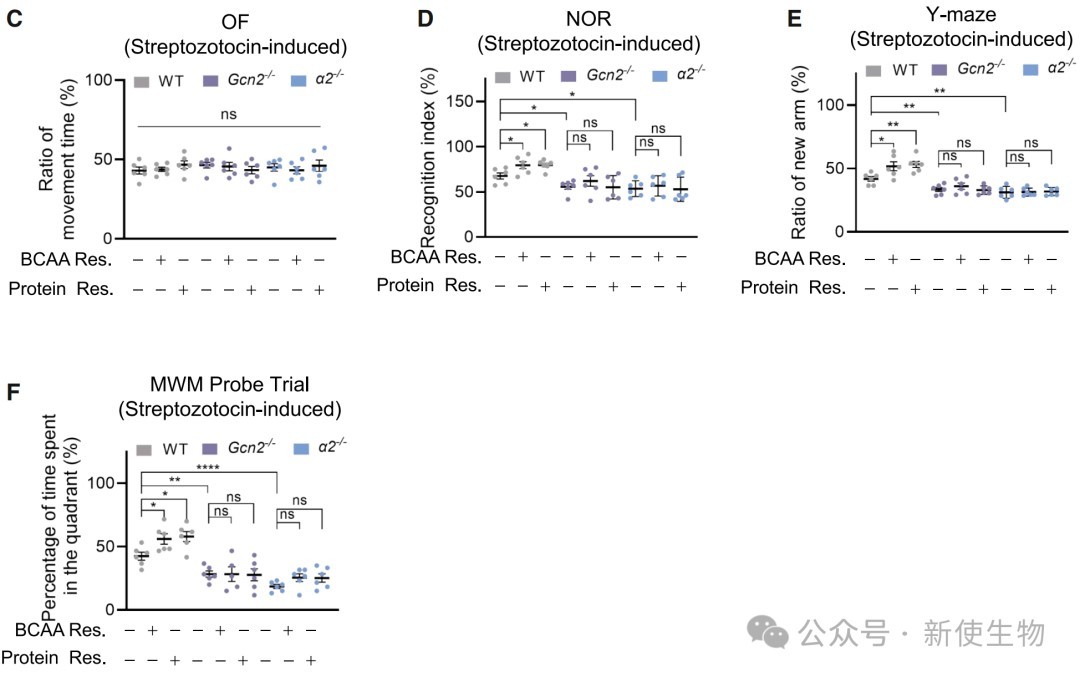
通过构建一系列基因敲除小鼠模型,研究发现敲除AMPKα2或GCN2,而非AMPKα1,会导致小鼠出现认知功能障碍、学习记忆能力下降等AD样症状。
这表明AMPKα2和GCN2在维持神经系统正常功能中扮演了α1所不具备的特异性角色。
三、AMPKα2-GCN2通路通过抑制蛋白质合成来响应氨基酸缺乏
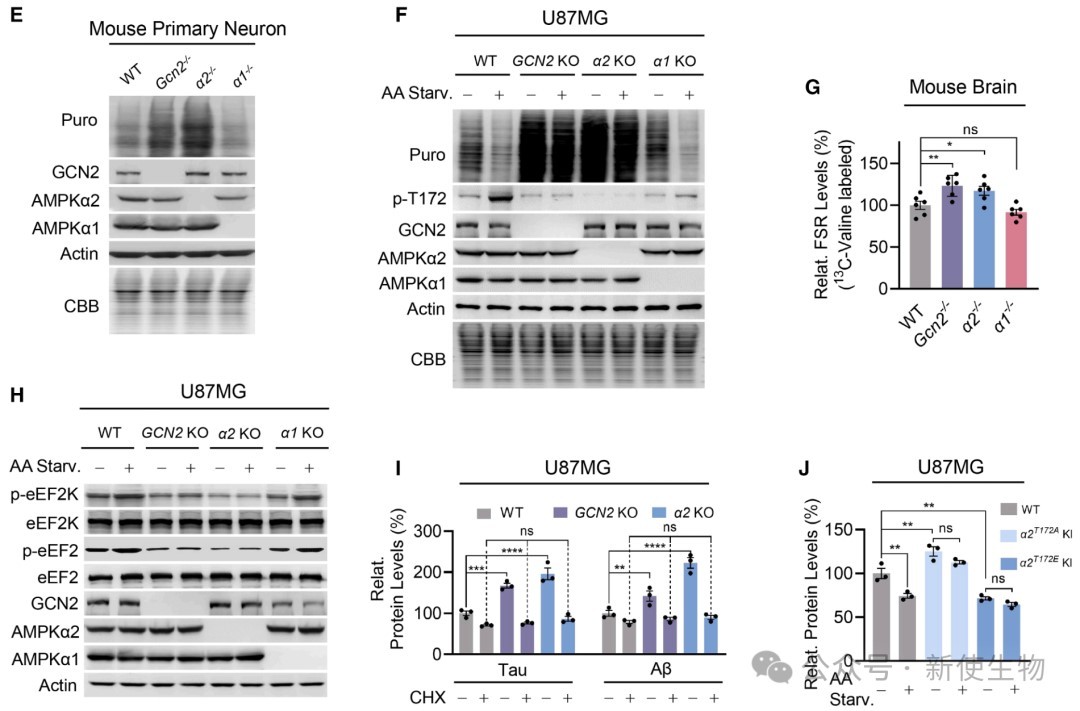
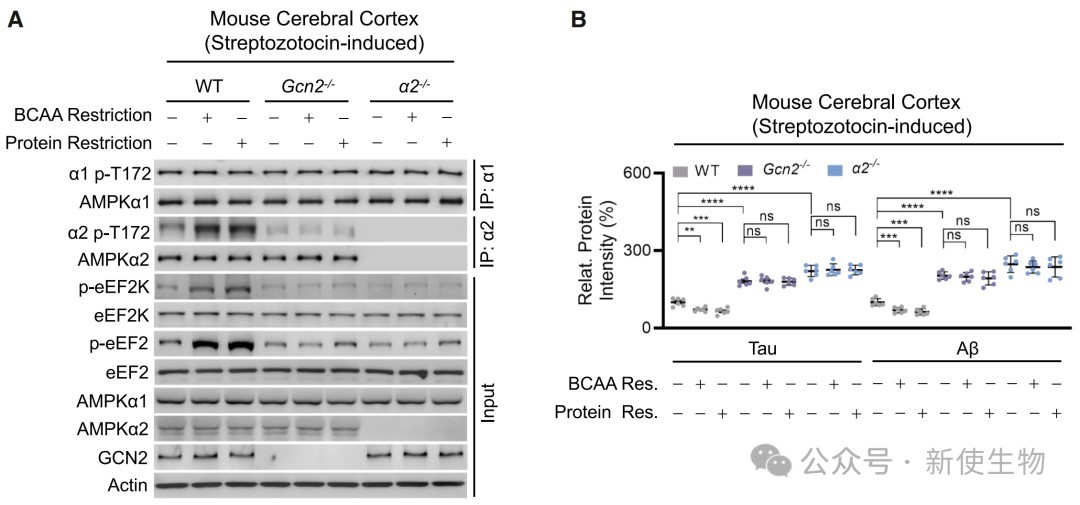
在细胞和小鼠模型中,敲除AMPKα2或GCN2会导致蛋白质合成速率增加和脑内氨基酸水平下降。
进一步研究发现,氨基酸缺乏能够诱导AMPKα2的T172位点磷酸化,进而抑制下游的翻译延伸因子eEF2,从而抑制蛋白质合成。
这一过程依赖于GCN2和α2,但独立于经典的AMPK能量感受信号。
四、GCN2是特异性磷酸化AMPKα2 T172位点的上游激酶
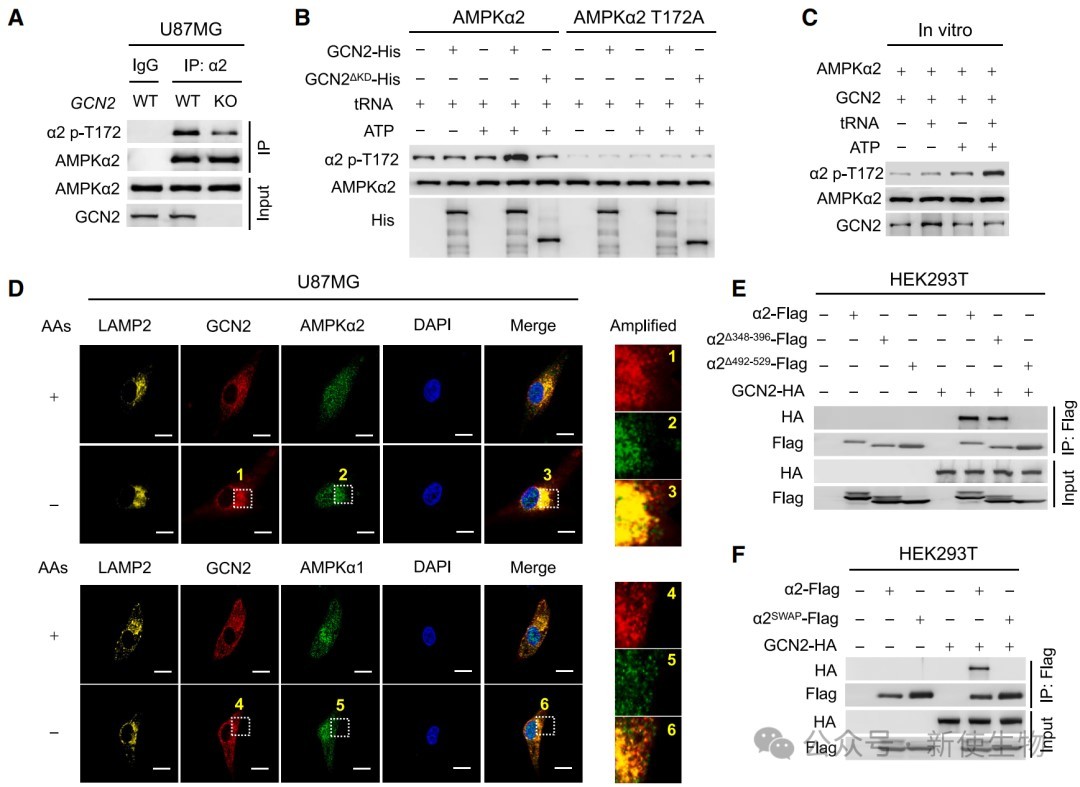
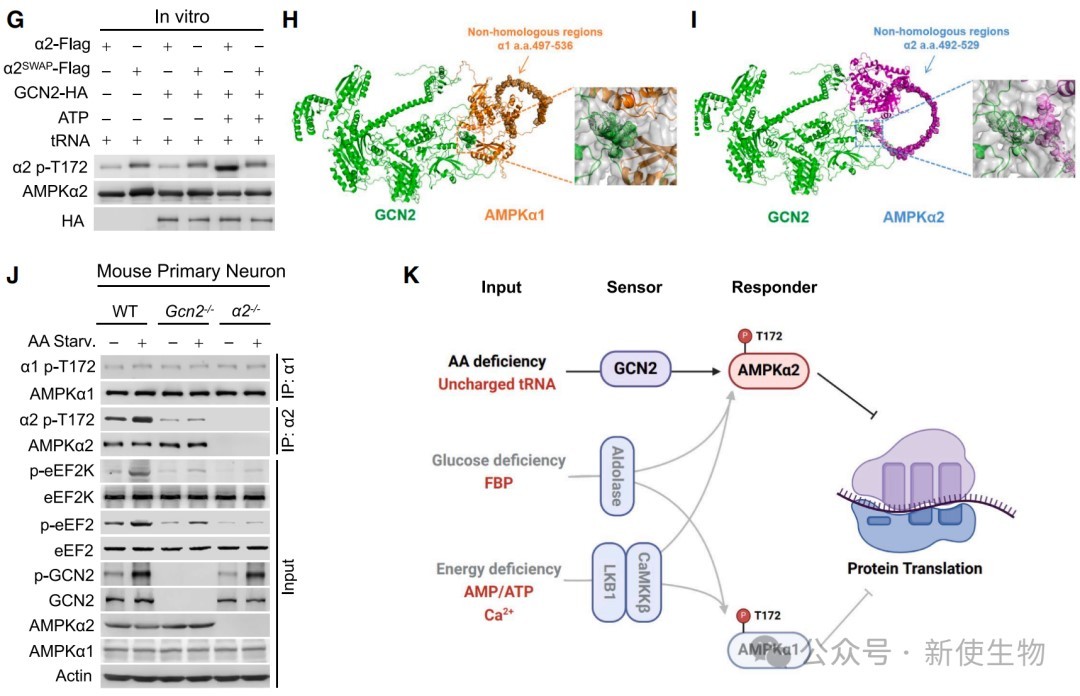
通过体外激酶实验和细胞内共定位分析,研究证实GCN2能够直接磷酸化AMPKα2的T172位点,而对α1无此作用。
这种特异性由α2蛋白上一段α1所没有的独特氨基酸序列(492-529区域)介导,该区域是GCN2与α2相互作用所必需的。
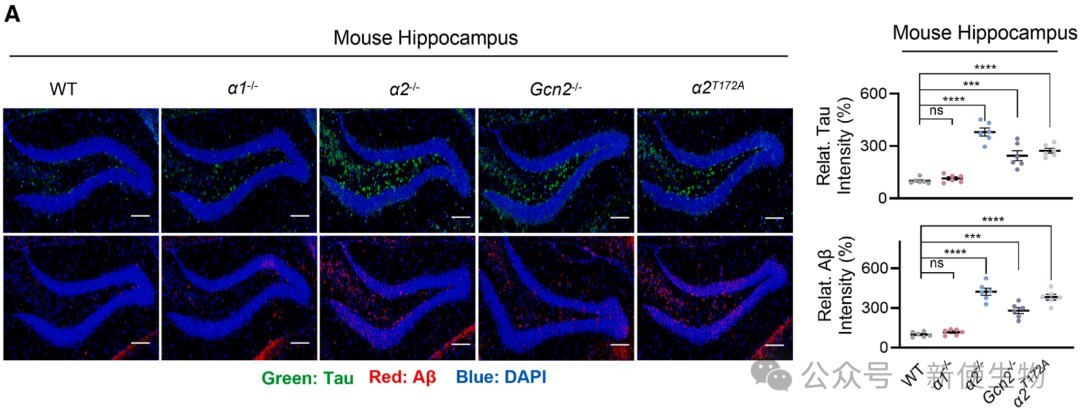
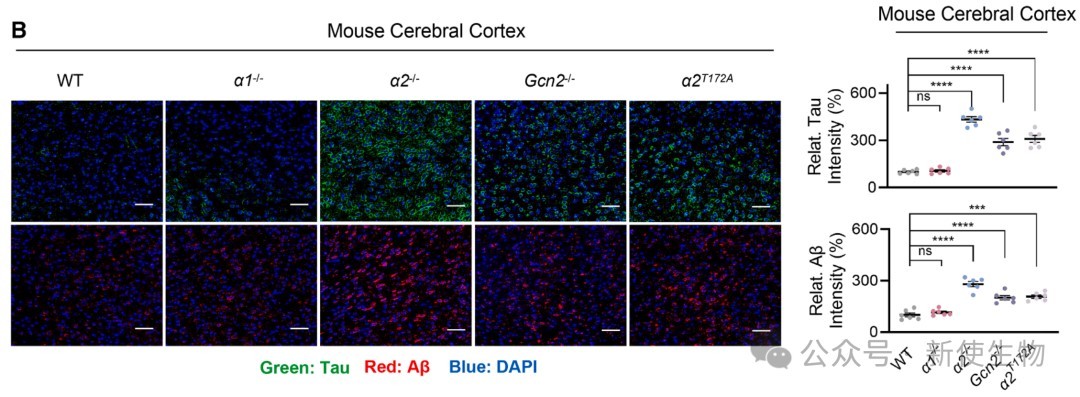
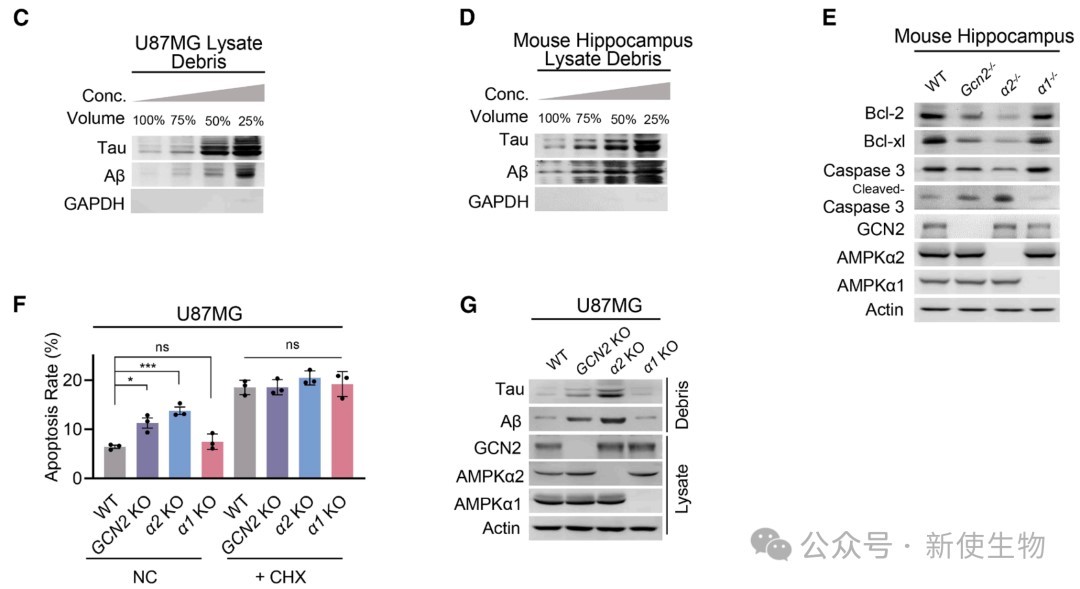
五、AMPKα2-GCN2通路失活导致病理性蛋白聚集和神经细胞凋亡
在敲除AMPKα2或GCN2的小鼠大脑中,作者观察到AD的标志性病理蛋白Tau和Aβ的显著聚集,以及神经细胞凋亡的增加。
这表明,该通路失活导致的蛋白质过度合成是诱发神经退行性病变的重要原因。
六、激活AMPK或限制饮食能够以α2依赖的方式改善AD症状
在AD小鼠模型中,使用AMPK激活剂(如二甲双胍)或进行饮食限制能够重新激活AMPKα2-T172磷酸化,抑制蛋白质过度合成,减少病理蛋白积累,并显著改善小鼠的认知功能。
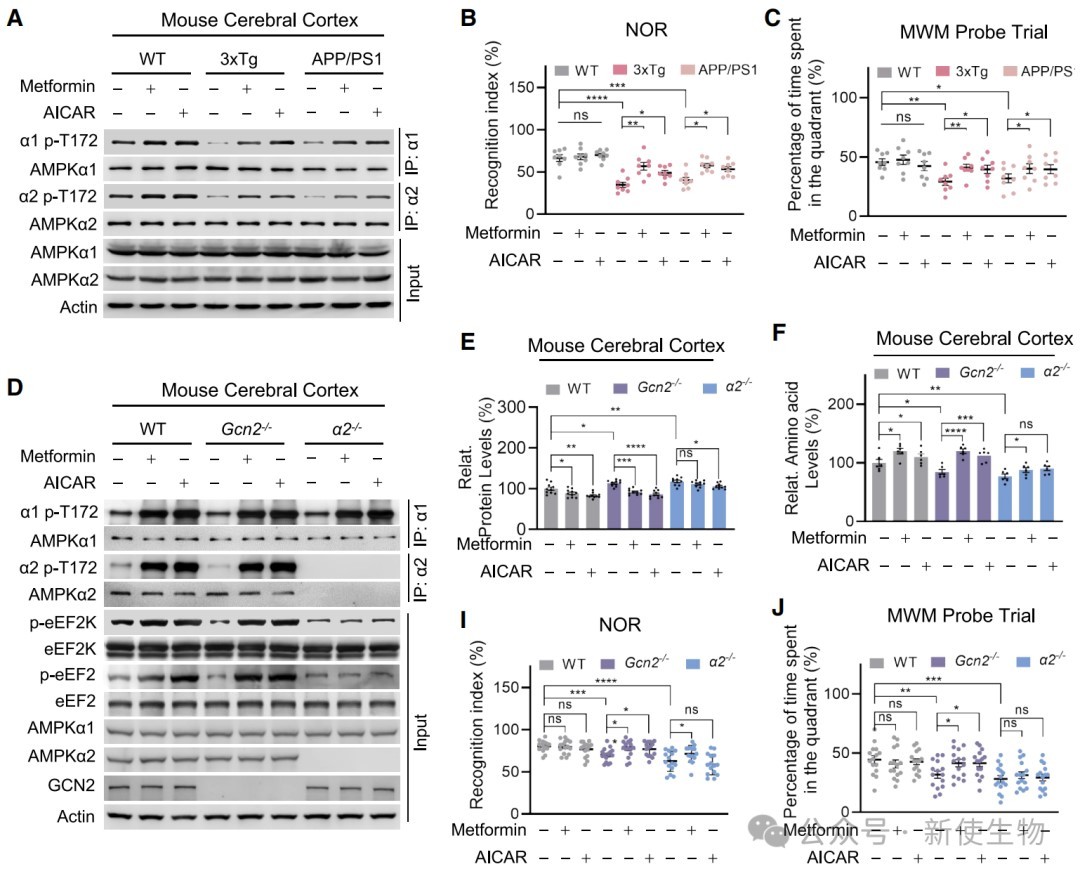
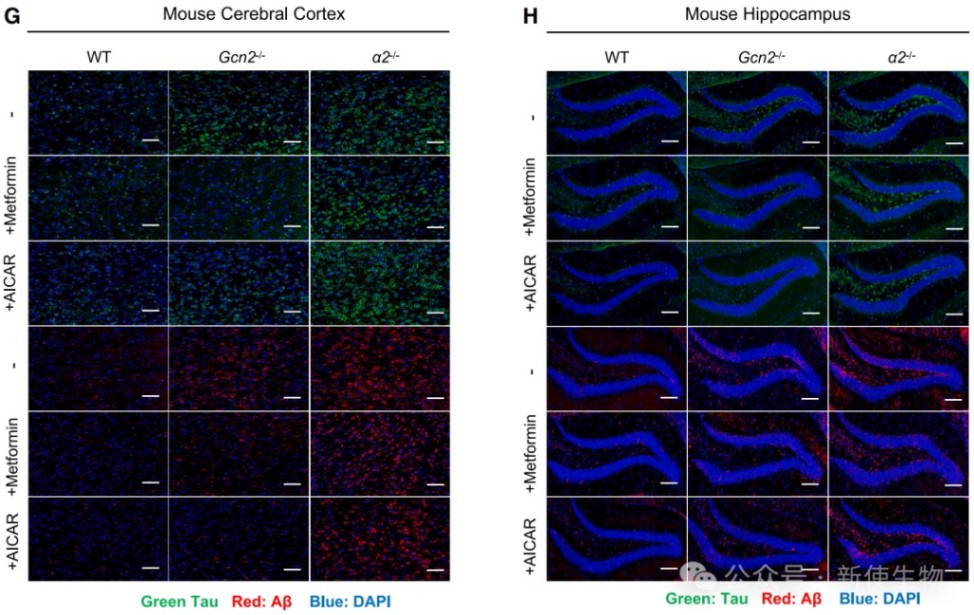
此外,这些有益效果在AMPKα2敲除小鼠中完全消失,证明了α2是这些干预措施发挥作用的关键靶点。
总结
本研究首次阐明了AMPK两个催化亚基α1和α2在功能上的特异性分工,鉴定出AMPKα2是感受氨基酸信号并调控蛋白质合成的关键分子。研究揭示了一条“氨基酸缺乏 → GCN2激活 → AMPKα2 T172磷酸化 → eEF2通路抑制 → 蛋白质合成减少”的全新信号通路,并证明该通路的失调是导致AD样病理和认知障碍的重要原因。该发现不仅深化了对AMPK功能多样性的理解,也为通过靶向AMPKα2或调节氨基酸摄入来预防和治疗AD等神经退行性疾病提供了新的理论依据和潜在策略。
| 新使生物专业翻译组一站式服务平台 |
| 产品名称 |
我们能够针对微量细胞或组织,如卵母细胞、卵巢、临床穿刺样品等产出高质量翻译组数据结果。
超高的准确性为研究非经典的开放阅读框(ORFs)提供极大便利,提高微肽(肿瘤新生抗原)的挖掘效率。
另外新使生物提供多物种多聚核糖体分析(Polysome profiling),了解更多翻译组技术信息可登录 www.neoribo.com。

点击图片查看
点击图片查看