首页
首页 关于我们
关于我们 产品中心
产品中心 技术服务
技术服务 技术中心
技术中心 联系我们
联系我们
导读
在真核细胞中,翻译终止由释放因子eRF1和eRF3识别mRNA上的终止密码子(UAA、UAG、UGA)来触发。但在特定情况下,近同源tRNA会抢先与终止密码子配对,使核糖体越过终止位点继续延伸,这一过程被称为翻译通读(TR)。
研究表明,TR并非随机发生的翻译错误,而是由顺式作用序列和反式作用因子共同定义的精密受控过程。然而,当基因突变产生提前终止密码子(PTC)时,会导致蛋白质翻译提前中断,产生截短且无功能的蛋白。
目前约有11%的人类遗传疾病归因于此类无义突变,包括囊性纤维化和杜氏肌营养不良症等。探索TR的分子机制并寻找能诱导PTC通读的药物,为治疗这类目前难以治愈的遗传病提供了潜在的药理学方案。
2026年3月24日,瑞士伯尔尼大学Oliver Mühlemann团队在Journal of Molecular Biology上发表了题为“Molecular Determinants and Therapeutic Targeting of Stop Codon Readthrough in Eukaryotic Translation”的深度综述。该文系统解析了决定终止密码子识别与通读效率的分子决定簇,详细探讨了增强PTC通读的小分子药物作用机制及临床开发前景,为治疗由无义突变引起的遗传性疾病提供了重要的理论指导。

综述整理
一、序列指纹:决定通读效率的“四碱基”逻辑
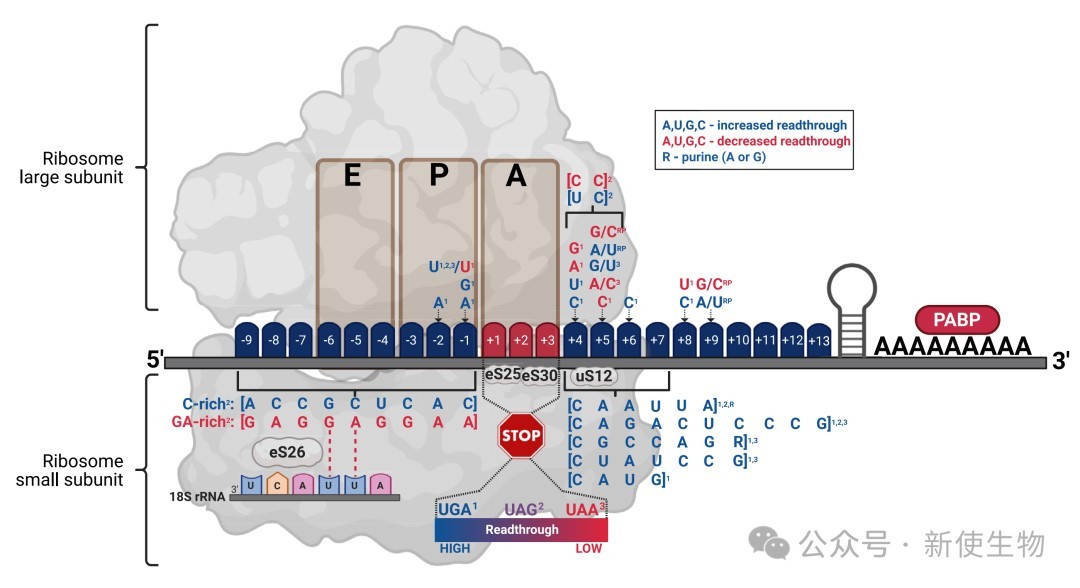
终止密码子的识别并非仅仅取决于三联体本身,而是由其周围的核苷酸序列(Sequence Context)协同定义的。
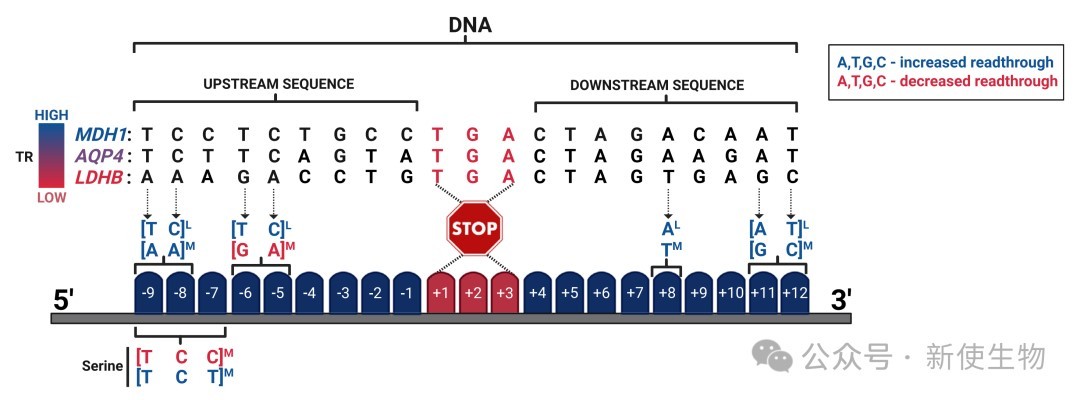
+4位碱基的核心作用:在核糖体解码中心,eRF1识别终止信号时涉及四个碱基。实验证明,紧随其后的+4位碱基对终止效率有决定性影响,其促进通读的能力遵循 C > U > A > G 的顺序。
终止密码子的层级:真核生物中三种终止密码子的通读易感性存在显著差异,UGA最容易发生通读,其次是UAG,而UAA则具有最高的翻译终止保真度。
上下游基序的调控:研究发现,通读效率还受到下游+5至+9位碱基,以及上游-1至-9位碱基的调节。例如,上游GA富集序列能增强终止,而C富集序列则易导致核糖体“滑动”越过终止点。
二、组织与衰老差异:通读活性的生理维度
翻译通读并非在所有细胞中以固定频率发生,它展现出显著的生理动态:
组织特异性:神经系统表现出最强的通读活性,这被认为有助于增加蛋白质组的多样性。相比之下,生殖系统的通读水平极低,以确保基因表达的高度精确。
衰老关联性:随着个体衰老,翻译保真度呈现下降趋势。在老年小鼠的肌肉和大脑中,观察到通读频率显著升高,这可能加剧了蛋白质稳态系统的崩溃,是衰老病理过程的一部分。
三、治疗策略:诱导PTC通读的小分子药理学
针对无义突变的药物开发主要聚焦于能够干扰核糖体解码精度的小分子:
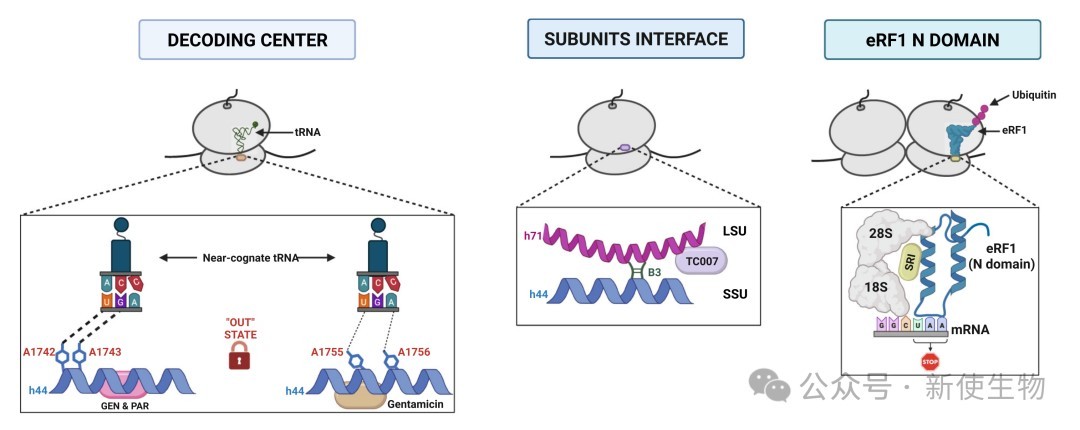
氨基糖苷类(Aminoglycosides):如Geneticin(G418)和庆大霉素。它们结合在核糖体解码中心,通过诱导A1742/A1743(18S rRNA)向外翻转,降低保真度阈值,使近同源tRNA能够错误配对并实现通读。
eRF1降解剂(SRI-41315):这是一种全新的机制,该类药物通过诱导释放因子eRF1的泛素化和蛋白酶体降解,降低其在细胞内的浓度,从而在PTC位点为tRNA竞争提供更大的窗口。
协同增敏(Mefloquine):抗疟药甲氟喹被发现能显著增强氨基糖苷类药物的通读活性,允许使用更低的药物浓度以减少耳毒性和肾毒性。
四、临床应用的挑战与未来方向
4.1) NMD效率的可变性是疗效的关键变量
无义介导的mRNA降解(NMD)通路通过降解含有PTC的mRNA来限制药物作用的底物,其效率的个体和组织差异是导致临床试验结果不一致的重要原因。
研究发现,NMD效率较低的患者对通读药物的响应更好。
4.2) 联合治疗:抑制NMD以增强通读疗效
在动物模型中,将NMD抑制剂与通读诱导剂联合使用,可产生1+1>2的协同效应。
通过稳定靶标mRNA的水平,显著增强了功能蛋白的恢复水平,为临床治疗提供了新思路。
4.3) 临床药物开发现状与展望
目前已有多种通读药物进入临床试验,如Ataluren(PTC124)和新一代氨基糖苷类衍生物ELX-02,后者在安全性及选择性上有了显著提升。
总结与展望
翻译通读绝非随机的翻译错误,而是一个受到mRNA局部序列上下文、细胞类型、年龄及反式作用因子高度动态调控的生物学过程。PTC与天然终止密码子在序列上下文、与翻译机器互作等方面存在根本差异,这为开发选择性靶向PTC的药物提供了理论基础。
未来的成功将不再依赖于“万金油”式的单一药物,而是需要建立一个包含多种药物的工具箱,并结合精准的基因组学分析,根据每位患者的具体突变及其上下文信息,来选择最合适的药物或药物组合,最终实现个性化的无义突变抑制疗法。
点击图片查看 点击图片查看新使生物专业翻译组一站式服务平台 产品名称